生物信息软件综合实践实验报告 实验题目 :表达谱基本分析及查询实验日期:2023年12月19日 星期二
实验者:生物信息2102 代子希
实验目的:
熟悉表达谱数据库的查询和数据下载
熟悉芯片表达谱数据分析的一般流程
掌握表达差异分析和基因富集分析的方法
了解常用的数据可视化方法
实验内容:
GEO数据库查询和数据下载
使用R包limma进行差异表达分析
使用R包clusterProfiler进行基因富集分析
使用gplots、ggpubr、pheatmap等R包对差异表达和富集分析进行结果可视化
实验流程和结果
以 GSE46456为例,该实验使用的芯片平台为GPL198,拟南芥样本基因型包括:野生型、BRI1 单突变型、GUL2单突变型、BRI和GUL 双突变型,每种基因型设置三种重复。研究三种突变型样本与WT野生型样本哪些基因存在显著的差异表达。根据所提供的演示代码和相关文件,请完成以下任务:
对获得的芯片数据进行数据标准化 、探针过滤 、limma差异分析 ,写明每一步骤的代码、目的以及中间结果。
运用limma获得突变体和野生型的差异表达基因集,并阐述差异分析结果的各列含义 。
对所有基因做GSEA富集分析;并对三组上调的差异表达基因 (bri1-WT、gul2-WT、bri1_gul2-WT)做GO富集分析,并解释富集结果,如有图片请注明图注信息。
安装、加载包以及相关依赖
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
# R ≥ 3.5.0
if ( ! requireNamespace ( "BiocManager" , quietly = TRUE ))
install.packages ( "BiocManager" )
BiocManager :: install ( version = "3.13" )
library ( BiocManager )
install.packages ( "cluster" )
install.packages ( "kohonen" )
install.packages ( "gplots" )
install.packages ( "ggpubr" )
install.packages ( "ggthemes" )
install.packages ( "pheatmap" )
BiocManager :: install ( "GEOquery" )
BiocManager :: install ( "RankProd" )
BiocManager :: install ( "affy" )
BiocManager :: install ( "affyPLM" )
BiocManager :: install ( "limma" )
BiocManager :: install ( "genefilter" )
BiocManager :: install ( "org.At.tair.db" )
BiocManager :: install ( "Mfuzz" )
BiocManager :: install ( "clusterProfiler" )
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
library ( RankProd )
library ( cluster )
library ( kohonen )
library ( gplots )
library ( RankProd )
library ( affy )
library ( affyPLM )
packageurl <- "https://bioconductor.org/packages//2.7/bioc/src/contrib/simpleaffy_2.64.0.tar.gz"
install.packages ( packageurl , repos = NULL , type = "source" )
library ( genefilter )
library ( gcrma )
options ( BioC_mirror = "https://mirrors.tuna.tsinghua.edu.cn/bioconductor" )
install.packages ( "https://github.com/SiYangming/simpleaffy/releases/download/v2.64.0/simpleaffy_2.64.0.tar.gz" , repos = NULL , type = "source" , dependencies = TRUE )
library ( simpleaffy )
library ( RColorBrewer )
library ( limma )
library ( pheatmap )
library ( Mfuzz )
library ( clusterProfiler )
library ( enrichplot )
library ( ggplot2 )
library ( "org.At.tair.db" , character.only = TRUE )
其中simpleaffy
使用源代码安装
1
2
packageurl <- "https://bioconductor.org/packages/3.8/bioc/src/contrib/simpleaffy_2.58.0.tar.gz"
install.packages(packageurl, repos=NULL, type="source")
下载数据
用 GEOquery 中 getGEOSuppFiles() 函数直接下载,指定下载路径
1
2
library ( GEOquery )
getGEOSuppFiles ( "GSE46456" , baseDir = "F:/tmp_data" )
报错
1
2
Error in getGEOSuppFiles ( "GSE46456" , baseDir = "F:/tmp_data" ) :
Failed to download F :/ tmp_data / GSE46456 / GSE46456_RAW.tar !
也许是网络原因,命令行使用代理也不行,直接到GEO数据库网页端下载芯片数据,在本地解压
1
2
3
4
#生成文件列表,以便批量导入文件
cels = list.files ( "./data/GSE46456/" , pattern = "*.gz" , full.names = TRUE )
# 读取CEL文件,将其处理成AffyBatch对象
celfiles <- ReadAffy ( filenames = cels )
数据预处理
1
2
3
4
5
6
7
8
9
10
11
# 将AffyBatch对象转换为ExpressionSet对象,对数据进行标准化
celfiles.rma <- rma ( celfiles )
Error in getCdfInfo ( object ) :
Could not obtain CDF environment , problems encountered :
Specified environment does not contain ATH1 -121501
Library - package ath1121501cdf not installed
Bioconductor - could not connect
此外: Warning message :
In readLines ( biocURL ) :
URL 'https://master.bioconductor.org/' : status was 'SSL connect error'
根据报错安装相关包
1
2
3
4
if ( ! requireNamespace ( "BiocManager" , quietly = TRUE ))
install.packages ( "BiocManager" )
BiocManager :: install ( "ath1121501cdf" )
1
2
3
4
# 将AffyBatch对象转换为ExpressionSet对象,对数据进行标准化
celfiles.rma <- rma ( celfiles )
#?rma
cols <- brewer.pal ( 8 , "Set1" )
1
2
3
4
5
6
? rma
rma { affy }
Robust Multi - Array Average expression measure
Description
This function converts an AffyBatch object into an ExpressionSet object using the robust multi - array average ( RMA ) expression measure.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
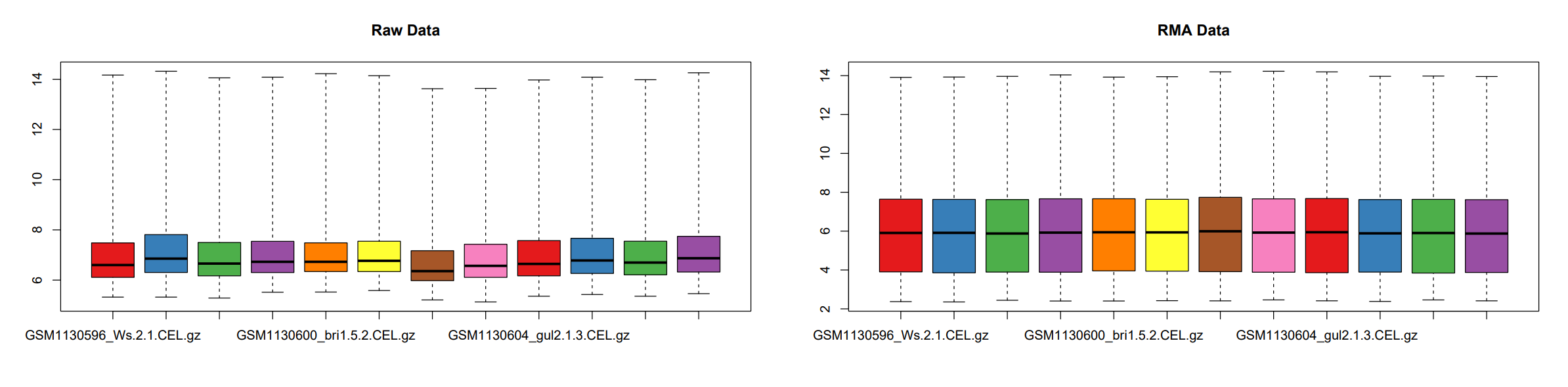
pdf ( "./png/boxplot_celfiles.pdf" )
boxplot ( celfiles , col = cols )
dev.off ()
pdf ( "./png/boxplot_celfiles_rma.pdf" )
boxplot ( celfiles.rma , col = cols )
dev.off ()
#放在一张图上,对比
pdf ( "./png/boxplot_celfiles_celfiles_rma.pdf" , width = 20 , height = 5 )
par ( mfrow = c ( 1 , 2 ))
boxplot ( celfiles , col = cols , main = "Raw Data" )
boxplot ( celfiles.rma , col = cols , main = "RMA Data" )
par ( mfrow = c ( 1 , 1 ))
dev.off ()
#密度和对数强度直方图
pdf ( "./png/hist_celfiles.pdf" )
hist ( celfiles , col = cols )
dev.off ()
pdf ( "./png/hist_celfiles_rma.pdf" )
hist ( celfiles.rma , col = cols )
dev.off ()
对比箱线图
对比标准化前后的数据,经过标准化后12个样本的数据分布更趋同
探针过滤
1
2
#对ExpressionSet的探针进行过滤(过滤掉表达量低的探针),返回一个list
celfiles.filtered <- nsFilter ( celfiles.rma , require.entrez = FALSE , remove.dupEntrez = FALSE )
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
#list中的eset为过滤后的ExpressionSet,filter.log为每一步过滤到多少探针的记录
> celfiles.filtered $ filter.log
$ numLowVar
[1] 11373
$ feature.exclude
[1] 64
> celfiles.filtered $ eset
ExpressionSet ( storageMode : lockedEnvironment )
assayData : 11373 features , 12 samples
element names : exprs
protocolData
sampleNames : GSM1130596_Ws -2-1 .CEL.gz GSM1130597_Ws -2-2 .CEL.gz ...
GSM1130607_gul2 -1 bri1 -5-3 .CEL.gz ( 12 total )
varLabels : ScanDate
varMetadata : labelDescription
phenoData
sampleNames : GSM1130596_Ws -2-1 .CEL.gz GSM1130597_Ws -2-2 .CEL.gz ...
GSM1130607_gul2 -1 bri1 -5-3 .CEL.gz ( 12 total )
varLabels : sample
varMetadata : labelDescription
featureData : none
experimentData : use 'experimentData(object)'
Annotation : ath1121501
1
2
#获得过滤后的表达矩阵
eset <- exprs ( celfiles.filtered $ eset )
表达量矩阵,每一行表示一个探针(基因),每一列为一个样本
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
> head ( eset )
GSM1130596_Ws -2-1 .CEL.gz GSM1130597_Ws -2-2 .CEL.gz
244901 _at 5.224648 5.428151
244902 _at 5.149407 5.187442
244903 _at 5.592680 5.436074
244904 _at 4.985820 5.072172
244906 _at 5.727308 5.889640
244912 _at 6.465566 6.586064
GSM1130598_Ws -2-3 .CEL.gz GSM1130599_bri1 -5-1 .CEL.gz
244901 _at 5.546510 4.683135
244902 _at 4.886097 4.672779
244903 _at 5.638751 5.562216
244904 _at 5.262937 5.016912
244906 _at 5.323069 5.381804
244912 _at 6.814510 7.653073
GSM1130600_bri1 -5-2 .CEL.gz GSM1130601_bri1 -5-3 .CEL.gz
244901 _at 4.753393 4.463033
244902 _at 4.805556 4.794880
244903 _at 5.622267 5.224591
244904 _at 5.446725 5.482161
244906 _at 5.609199 5.514687
244912 _at 7.871753 8.260488
GSM1130602_gul2 -1-1 .CEL.gz GSM1130603_gul2 -1-2 .CEL.gz
244901 _at 6.087000 5.868863
244902 _at 5.527636 5.592619
244903 _at 6.605356 6.062327
244904 _at 5.366306 5.495490
244906 _at 6.348055 6.080350
244912 _at 8.052288 7.970124
GSM1130604_gul2 -1-3 .CEL.gz GSM1130605_gul2 -1 bri1 -5-1 .CEL.gz
244901 _at 5.386404 5.490967
244902 _at 5.927320 5.302640
244903 _at 5.597098 6.694592
244904 _at 5.026725 5.257816
244906 _at 6.218219 6.613726
244912 _at 7.871955 7.620157
GSM1130606_gul2 -1 bri1 -5-2 .CEL.gz GSM1130607_gul2 -1 bri1 -5-3 .CEL.gz
244901 _at 5.671008 5.305481
244902 _at 5.524203 5.494279
244903 _at 6.661931 6.565135
244904 _at 5.356717 5.228070
244906 _at 6.537190 6.425924
244912 _at 7.387385 7.555005
> colnames ( eset )
[1] "GSM1130596_Ws-2-1.CEL.gz" "GSM1130597_Ws-2-2.CEL.gz"
[3] "GSM1130598_Ws-2-3.CEL.gz" "GSM1130599_bri1-5-1.CEL.gz"
[5] "GSM1130600_bri1-5-2.CEL.gz" "GSM1130601_bri1-5-3.CEL.gz"
[7] "GSM1130602_gul2-1-1.CEL.gz" "GSM1130603_gul2-1-2.CEL.gz"
[9] "GSM1130604_gul2-1-3.CEL.gz" "GSM1130605_gul2-1bri1-5-1.CEL.gz"
[11] "GSM1130606_gul2-1bri1-5-2.CEL.gz" "GSM1130607_gul2-1bri1-5-3.CEL.gz"
探针注释
注释探针: 防止非特异性结合造成的干扰(会有多个探针检测同一个基因的表达)。
先对探针进行注释,确定每个探针对应检测哪个基因的表达,然后再合并重复探针
1
2
3
4
5
ara_anno <- read.delim ( "./data/affy_ATH1_array_elements-2010-12-20.txt" )
ids <- match ( rownames ( eset ), ara_anno $ array_element_name )
rownames ( eset ) <- ara_anno $ locus[ids]
colnames ( eset ) <- sub ( ".CEL.gz" , "" , colnames ( eset ))
规范矩阵的行名和列名
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
> head ( eset )
GSM1130596_Ws -2-1 GSM1130597_Ws -2-2 GSM1130598_Ws -2-3
ATMG00640 5.224648 5.428151 5.546510
ATMG00650 5.149407 5.187442 4.886097
ATMG00660 5.592680 5.436074 5.638751
ATMG00670 4.985820 5.072172 5.262937
ATMG00690 5.727308 5.889640 5.323069
AT2G07783 ; ATMG00830 6.465566 6.586064 6.814510
GSM1130599_bri1 -5-1 GSM1130600_bri1 -5-2 GSM1130601_bri1 -5-3
ATMG00640 4.683135 4.753393 4.463033
ATMG00650 4.672779 4.805556 4.794880
ATMG00660 5.562216 5.622267 5.224591
ATMG00670 5.016912 5.446725 5.482161
ATMG00690 5.381804 5.609199 5.514687
AT2G07783 ; ATMG00830 7.653073 7.871753 8.260488
GSM1130602_gul2 -1-1 GSM1130603_gul2 -1-2 GSM1130604_gul2 -1-3
ATMG00640 6.087000 5.868863 5.386404
ATMG00650 5.527636 5.592619 5.927320
ATMG00660 6.605356 6.062327 5.597098
ATMG00670 5.366306 5.495490 5.026725
ATMG00690 6.348055 6.080350 6.218219
AT2G07783 ; ATMG00830 8.052288 7.970124 7.871955
GSM1130605_gul2 -1 bri1 -5-1 GSM1130606_gul2 -1 bri1 -5-2
ATMG00640 5.490967 5.671008
ATMG00650 5.302640 5.524203
ATMG00660 6.694592 6.661931
ATMG00670 5.257816 5.356717
ATMG00690 6.613726 6.537190
AT2G07783 ; ATMG00830 7.620157 7.387385
GSM1130607_gul2 -1 bri1 -5-3
ATMG00640 5.305481
ATMG00650 5.494279
ATMG00660 6.565135
ATMG00670 5.228070
ATMG00690 6.425924
AT2G07783 ; ATMG00830 7.555005
limma差异分析
构建分组矩阵
1
2
3
4
5
group_list = c ( rep ( "wt" , 3 ), rep ( "bri1" , 3 ), rep ( "gul2" , 3 ), rep ( "gul_bri" , 3 ))
design <- model.matrix ( ~ 0 + factor ( group_list ))
colnames ( design ) <- levels ( factor ( group_list ))
rownames ( design ) <- colnames ( eset )
构建分组矩阵,和野生型作比较
1
2
contrast.matrix <- makeContrasts ( bri1 - wt , gul2 - wt , gul_bri - wt , levels = design )
contrast.matrix
limma三部曲:线性模型拟合 根据对比模型进行差值计算 贝叶斯检验
1
2
3
4
#limma"三部曲"
fit1 <- lmFit ( eset , design ) #线性模型拟合
fit2 <- contrasts.fit ( fit1 , contrast.matrix ) #根据对比模型进行差值计算
fit2 <- eBayes ( fit2 ) #贝叶斯检验
输出差异表达基因
1
2
3
4
5
6
7
8
9
10
11
#利用toptable 导出DEG结果
limma_results <- lapply ( colnames ( contrast.matrix ), function ( x ){ topTable ( fit2 , coef = x , adjust = "fdr" , sort.by = "logFC" , number = Inf )})
length ( limma_results )
names ( limma_results ) <- colnames ( contrast.matrix ) #对导出的结果标记title信息
head ( limma_results[[1]] )
save ( limma_results , file = "./data/limma_compare_res.RData" )
#对每对比较的样本对DEG结果单独导出DEG信息6
for ( n in names ( limma_results )){
write.table ( limma_results[[n]] , file = sprintf ( "%s.tsv" , paste0 ( "./data/" , gsub ( " " , "" , n ))), row.names = FALSE , sep = "\t" )
}
save ( eset , file = "./data/eset.RData" )
差异分析结果解释
一共有三组结果,去循环最后一个文件查看
1
2
3
4
5
> n
[1] "gul_bri - wt"
> colnames ( limma_results[[n]] )
[1] "ID" "logFC" "AveExpr" "t" "P.Value" "adj.P.Val"
[7] "B"
1
2
3
4
5
6
7
8
> head ( limma_results[[n]] )
ID logFC AveExpr t P.Value adj.P.Val B
2773 AT5G12030 4.279782 8.119452 41.36505 1.465057e-20 1.666210e-16 37.01501
9250 AT2G40170 4.106650 7.424729 39.00006 4.589678e-20 2.609920e-16 35.95876
1669 AT5G54190 3.931407 8.501771 35.62676 2.644989e-19 5.013576e-16 34.31203
6603 AT3G15670 3.917131 7.540334 35.89093 2.292873e-19 5.013576e-16 34.44747
9416 AT2G21490 3.909309 6.788934 31.58778 2.697592e-18 2.045314e-15 32.08537
46 ATCG00790 3.901580 10.302096 34.51467 4.882151e-19 7.932100e-16 33.72882
ID : Gene IDlogFC : 两组表达值之间以2为底对数化的变化倍数(Fold change, FC),由于基因表达矩阵本身已经取了对数,这里是两组基因表达值均值之差AveExpr :该探针组所在所有样品中的平均表达值t :贝叶斯调整后的两组表达值间 T 检验中的 t 统计量P.Value : 检验P值adj.P.Val :调整后的 P 值(多重检验BH等方法)B :是经验贝叶斯得到的标准差的对数化值
差异表达分析结果可视化
热图
1
2
3
pdf ( "./png/heatmap.pdf" )
pheatmap ( eset , col = c ( colorRampPalette ( brewer.pal ( 9 , "Blues" ) [7 : 2 ] )( 100 ), colorRampPalette ( brewer.pal ( 9 , "Reds" ) [2 : 7 ] )( 100 )), border_color = NA , cluster_rows = T , cluster_cols = T , show_rownames = F , show_colnames = T , angle_col = 315 , fontsize = 13 , main = "expression" , display_numbers = F )
dev.off ()
颜色的深浅来反映数值的高低,聚类反映不同基因和不同样本之间的表达量相似性
火山图
1
2
3
4
5
6
7
8
9
10
11
12
13
14
library(ggpubr)
library(ggthemes)
for (n in names(limma_results)){
file_name = sprintf("%s.tsv",paste0("./data/", gsub(" ","",n)))
deg.data <- read.table(file_name,header=T, sep="\t")
deg.data$logP <- -log10(deg.data$adj.P.Val)
deg.data$Group = "not-significant" #定义Group列
deg.data$Group[which ((deg.data$adj.P.Val < 0.05) & (deg.data$logFC > 2))] = "up-regulated" #定义DEG标准
deg.data$Group[which ((deg.data$adj.P.Val < 0.05) & (deg.data$logFC < -2))] = "down-regulated" #定义DEG标准
table(deg.data$Group)
pdf(sprintf("./png/%s.pdf",paste0(gsub(" ","",n))))
ggscatter(deg.data, x="logFC",y = "logP",color = "Group") + theme_base() + labs(title = n)
dev.off()
}
1
2
3
4
5
6
7
8
9
10
11
12
13
14
#新加一列lable
deg.data $ Lable = ""
#对差异表达基因P值从小到大排序
deg.data <- deg.data [order ( deg.data $ adj.P.Val ), ]
#从高表达基因中选取adj.P.Val最显著的10个基因
up.genes <- head ( deg.data $ ID [which ( deg.data $ Group == "up-regulated" ) ] , 10 )
#从低表达基因中选取adj.P.Val最显著的10个基因
down.genes <- head ( deg.data $ ID [which ( deg.data $ Group == "down-regulated" ) ] , 10 )
# 讲上两步选取的显著基因合并并加入到lable中
deg.top10.genes <- c ( as.character ( up.genes ), as.character ( down.genes ) )
deg.data $ Lable [match ( deg.top10.genes , deg.data $ ID ) ] <- deg.top10.genes
png ( "./png/plot2.png" , res = 300 , width = 10 , height = 10 , units = "in" )
ggscatter ( deg.data , x = "logFC" , y = "logP" , color = "Group" , palette = c ( "#2f5688" , "#BBBBBB" , "#CC0000" ), size = 1 , label = deg.data $ Lable , font.label = 8 , repel = T , xlab = "Log2FoldChange" , ylab = "-Log10(Adjust P-value)" ,) + theme_base () + geom_hline ( yintercept = 1.30 , linetype = "dashed" ) + geom_vline ( xintercept = c ( -2 , 2 ), linetype = "dashed" )
dev.off ()
富集分析
首先进行GSEA
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
for ( n in names ( limma_results )){
file_name <- sprintf ( '%s.tsv' , paste0 ( "./data/" , gsub ( " " , "" , n )))
GSEA_data <- read.table ( file_name , header = T , sep = "\t" )
GSEA_gene_lists <- GSEA_data $ logFC #提取表达量变化值
names ( GSEA_gene_lists ) <- GSEA_data $ ID #给提取出来的值赋予ID
GSEA_gene_lists <- sort ( GSEA_gene_lists , decreasing = TRUE ) #降序排列
organisms <- get ( "org.At.tair.db" ) #获取拟南芥数据库信息
#GSEA富集分析
GSEA_GO_Result <-
gseGO (
geneList = GSEA_gene_lists ,
ont = "ALL" ,
keyType = "TAIR" ,
nPerm = 10000 ,
minGSSize = 3 ,
maxGSSize = 800 ,
pvalueCutoff = 0.05 ,
verbose = TRUE ,
OrgDb = organisms ,
pAdjustMethod = "none"
)
Sort_GO_result <- GSEA_GO_Result [order ( GSEA_GO_Result $ enrichmentScore , decreasing = T ), ]
write.table ( Sort_GO_result , paste ( file_name , 'GSEA_GO_Result.txt' , sep = '_' ), sep = "\t" , quote = F , col.names = T , row.names = F )
GSEA_GO_Result_plot <- gseaplot2 ( GSEA_GO_Result , row.names ( Sort_GO_result ) [1 : 4 ] , title = n )
png ( filename = paste ( file_name , 'GSEA_GO_Result.png' , sep = '_' ), width = 3580 , height = 2200 , res = 300 )
print ( GSEA_GO_Result_plot )
dev.off ()
GSEA_GO_Result_plot2 <- gseaplot ( GSEA_GO_Result , by = "all" , title = GSEA_GO_Result $ Description[1] , geneSetID = 1 )
png ( filename = paste ( file_name , 'GSEA_GO_Result2.png' , sep = '_' ), width = 3580 , height = 2200 , res = 300 )
print ( GSEA_GO_Result_plot2 )
dev.off ()
}
通过 row.names(Sort_GO_result)[1:4]指定每一组中富集分数最高的四个term画富集条码图
对三组上调的差异表达基因(bri1-WT、gul2-WT、bri1_gul2-WT)做GO富集分析
1
2
3
4
5
6
7
8
9
10
11
12
13
14
for ( n in names ( limma_results )){
file_name <- sprintf ( '%s.tsv' , paste0 ( "./data/" , gsub ( " " , "" , n )))
deg.data <- read.table ( file_name , header = T , sep = "\t" )
deg.data $ logP <- - log10 ( deg.data $ adj.P.Val ) #-log10值转换
deg.data $ Group = "not-significant" #定义Group列
deg.data $ Group [which (( deg.data $ adj.P.Val < 0.05 ) & ( deg.data $ logFC > 2 )) ] = "up-regulated" #定义DEG标准
deg.data $ Group [which (( deg.data $ adj.P.Val < 0.05 ) & ( deg.data $ logFC < -2 )) ] = "down-regulated" #定义DEG标准
data <- deg.data[deg.data $ Group == "up-regulated" , ]
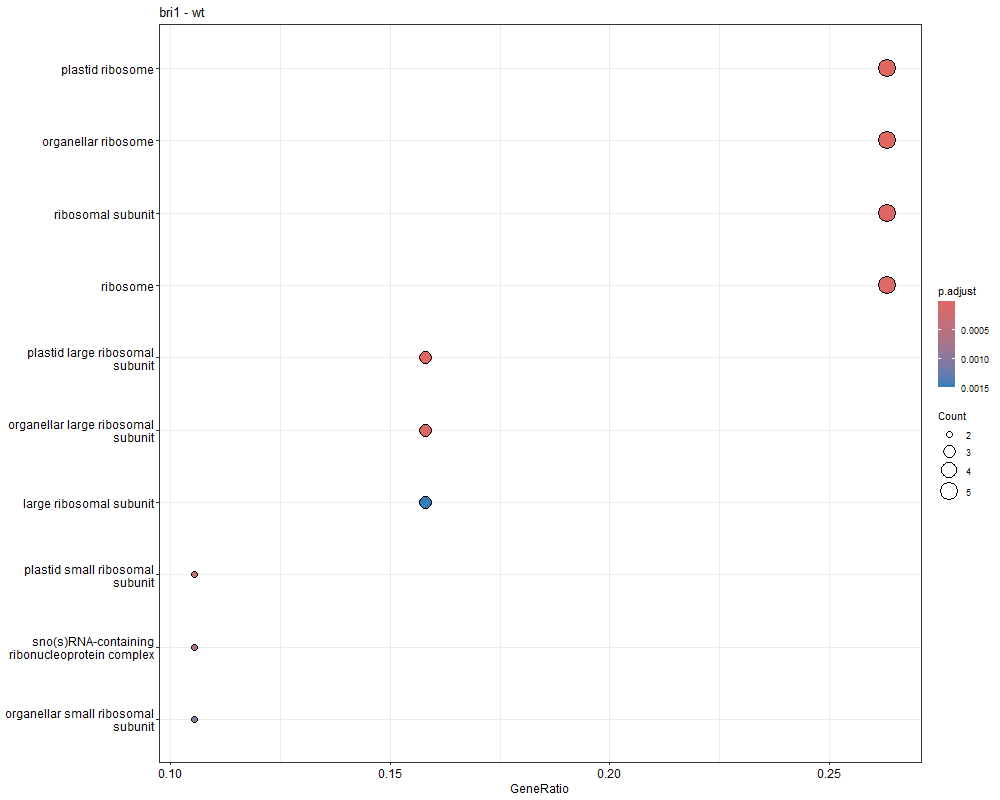
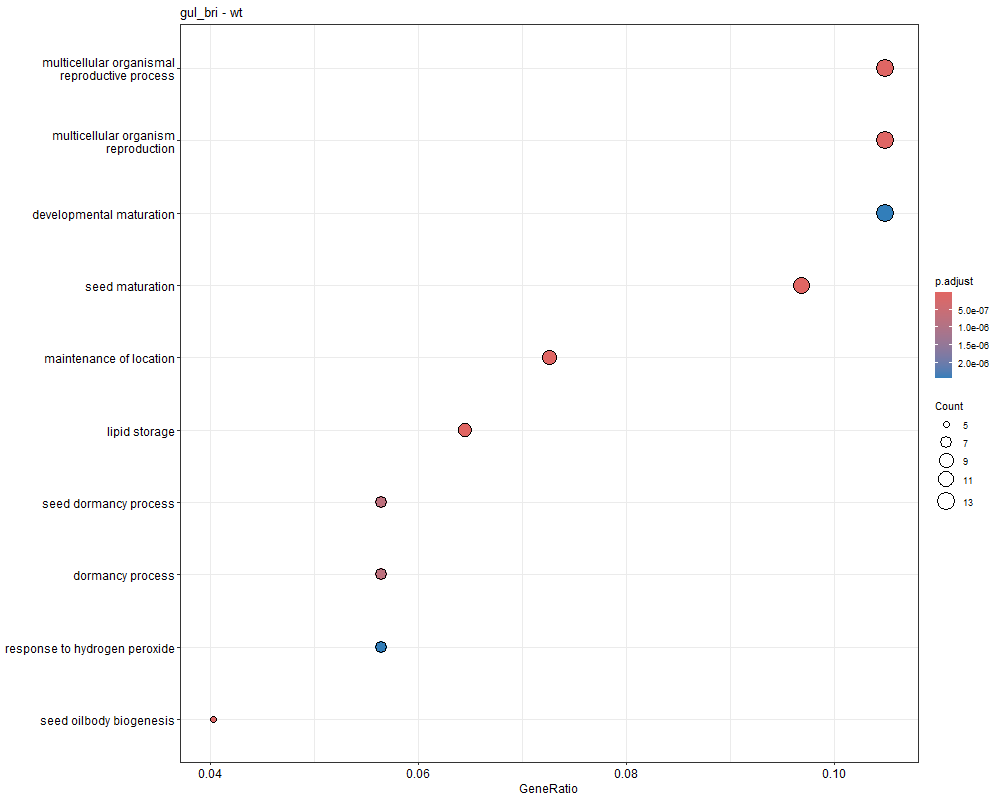
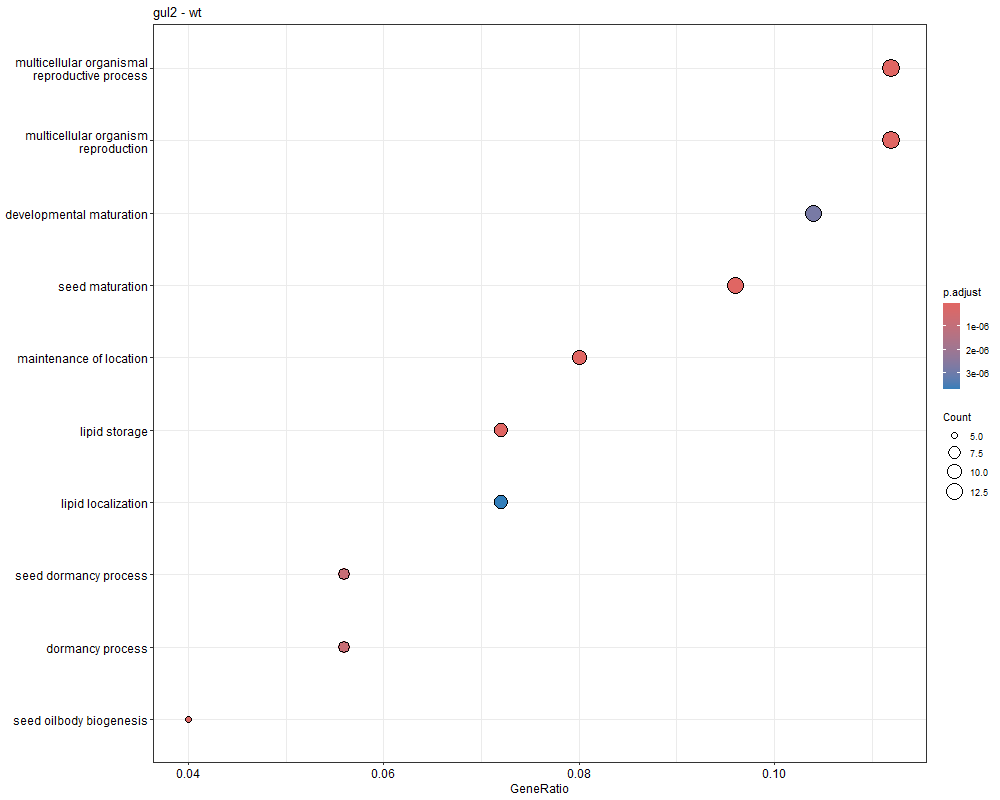
ego <- enrichGO ( gene = data $ ID , keyType = "TAIR" , OrgDb = organisms , ont = "ALL" , pAdjustMethod = "BH" , qvalueCutoff = 0.05 )
png ( file = paste ( file_name , 'GO_dot.png' , sep = '_' ), bg = "transparent" , width = 1000 , height = 800 )
GO_dot <- dotplot ( ego , showCategory = 10 ) + ggtitle ( n )
print ( GO_dot )
dev.off ()
}
bri1-WT GO富集分析
gul2-WT GO富集
bri1_gul2-WT GO富集
思考与讨论
1.为什么要对探针信号或基因表达量取对数?为什么是log2不是log10或lg?
在芯片数据分析中,对探针信号或基因表达量取对数是为了减小数据的离散程度,使得数据更加符合正态分布。取对数后,数据的性质和相对关系不会受到影响。
用log2,因为基因表达量增加1倍被认为就可以造成生物学上的一些变化,而且如果前面是对表达量取log2,后面计算fold-chage就可以直接相减。
2.为什么要设置生物学重复?
由于遗传和环境等因素的影响会引起有机体的个体差异,因此需要采用生物重复的实验设计方法来消除该差异。对同一个处理组中独立来源的多个样本分别进行独立测定分析,是整个实验的完全重复。设计生物重复可以:
能够消除组内误差:生物学重复可以测量变异程度;
增强结果的可靠性:测序的样本数越多,越能够降低背景差异;
检测离群样本:异常样本的存在,会严重影响测序结果的准确性,通过计算样本间的相关性可以发现异常样本,将其排除。
3.芯片数据差异表达分析和富集分析中,分别需要用到哪些算法、统计模型和检验?
在芯片数据差异表达分析中,常用的算法和统计模型包括:
倍数分析方法:倍数变换fold change,单纯的case与control组表达值相比较,对没有重复实验样本的芯片数据,或者双通道数据采用这种方法。
参数法分析(t检验):当t超过根据可信度选择的标准时, 比较的两样本被认为存在着差异。但小样本基因芯片实验会导致不可信的变异估计,此时采用调节性T检验。
非参数分析:由于微阵列数据存在“噪声”干扰而且不满足正态分布假设,用t检验有风险。非参数检验并不要求数据满足特殊
在富集分析中,常用的统计方法包括累计超几何分布和Fisher精确检验。由于在进行富集分析时通常需要同时进行大量检验(多重检验),所以需要采用多重检验校正的方法对检验结果进行校正,常用的校正方法包括Bonferroni校正和Benjiamini false discovery rate校正。