利用ChIPseeker R包对peak进行注释
经过前期的处理拿到了BED文件,利用这些文件和ChIPSeeker可以进一步对peak注释、可视化。
指定Bioregion结合图谱可视化
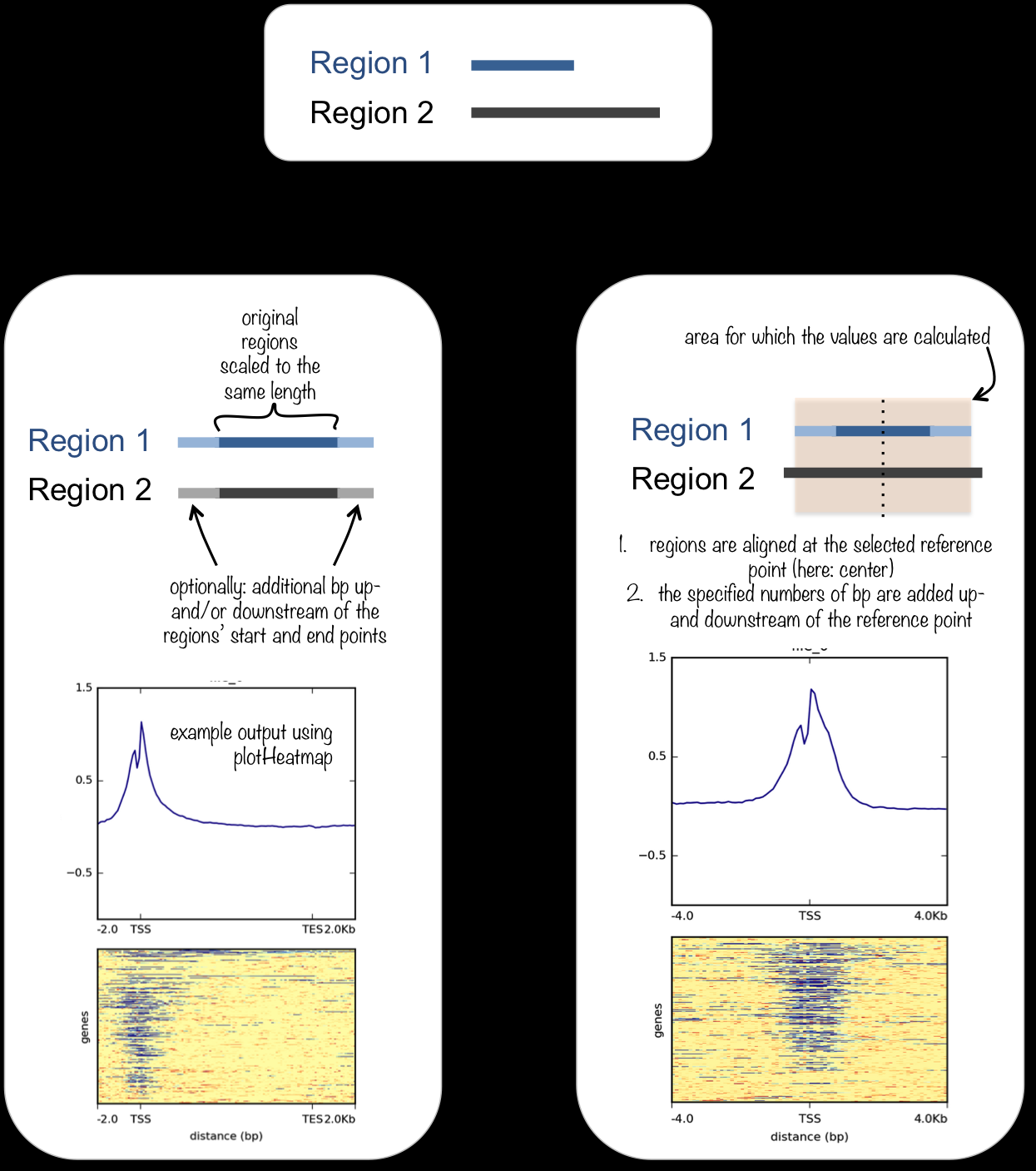
网上大部分教程都是限定一个固定的窗口(比如最常用到的TSS启动子区域),将所有的peak全部align(标准化对齐?),可视化其结合谱图。而我想要的效果是 将**整个基因(从TSS上游限定部分至TTS下游限定部分)**作为一个窗口
类似效果如下图左侧的 original regions scaled to the same length
 deeptools示例
deeptools示例
经过一系列bing搜索找到了这个问题的解决顺寻:
could the "plotAvgProf" can get the region from TSS to TTS ? · Issue #87 · YuLab-SMU/ChIPseeker · GitHu
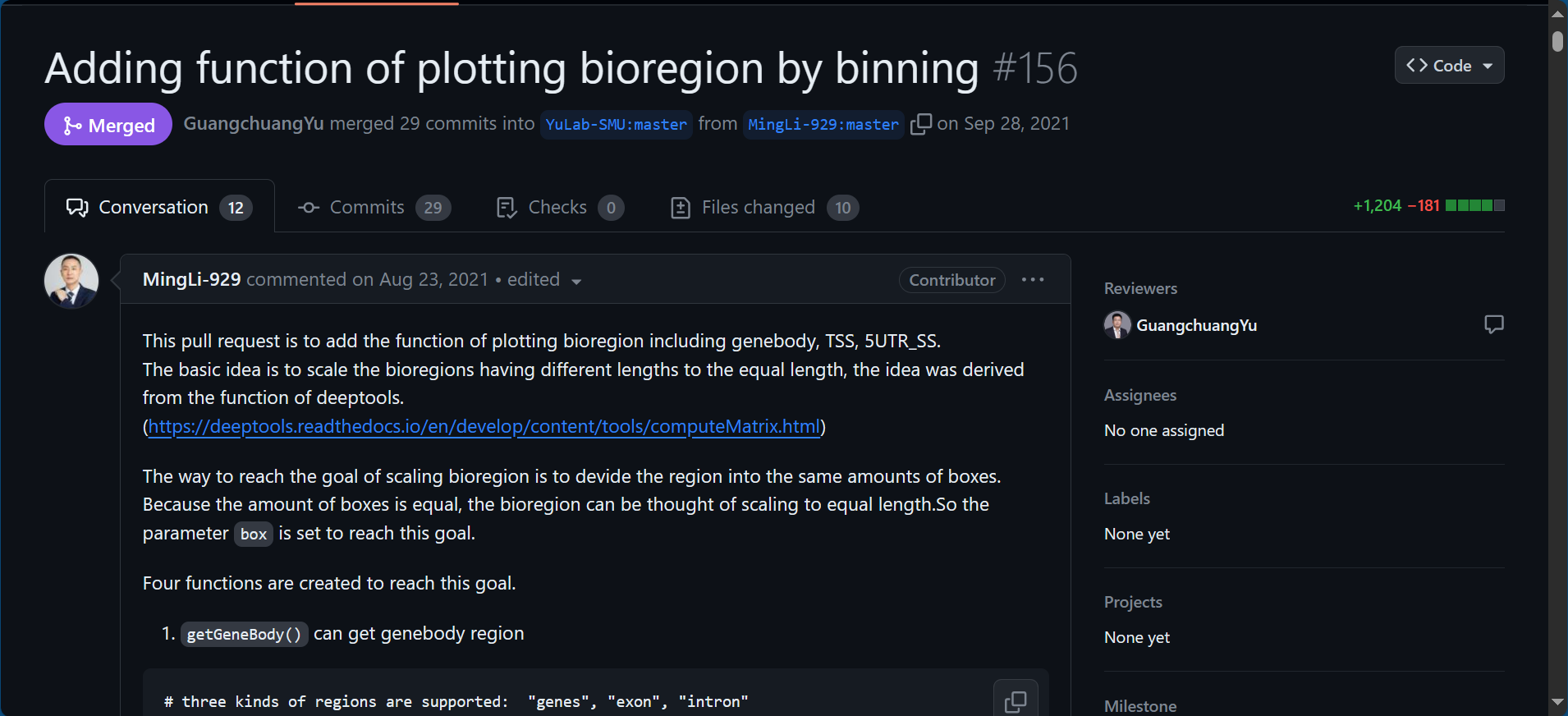
Adding function of plotting bioregion by binning by MingLi-929 · Pull Request #156 · YuLab-SMU/ChIPseeker (github.com)
Adding function of plotting bioregion by binning #156
在这个界面中,作者借鉴deeptools中computeMatrix的分bin方法归一化不同BioRegion的长度,加入了一系列函数实现上述设想目的。
但是实践后发现在最新的包里面似乎没有了这些函数,最后在bioconductor中找到一些详细的示例说明,发现函数名称功能重整。
ChIPseeker: an R package for ChIP peak Annotation, Comparison and Visualization (bioconductor.org)
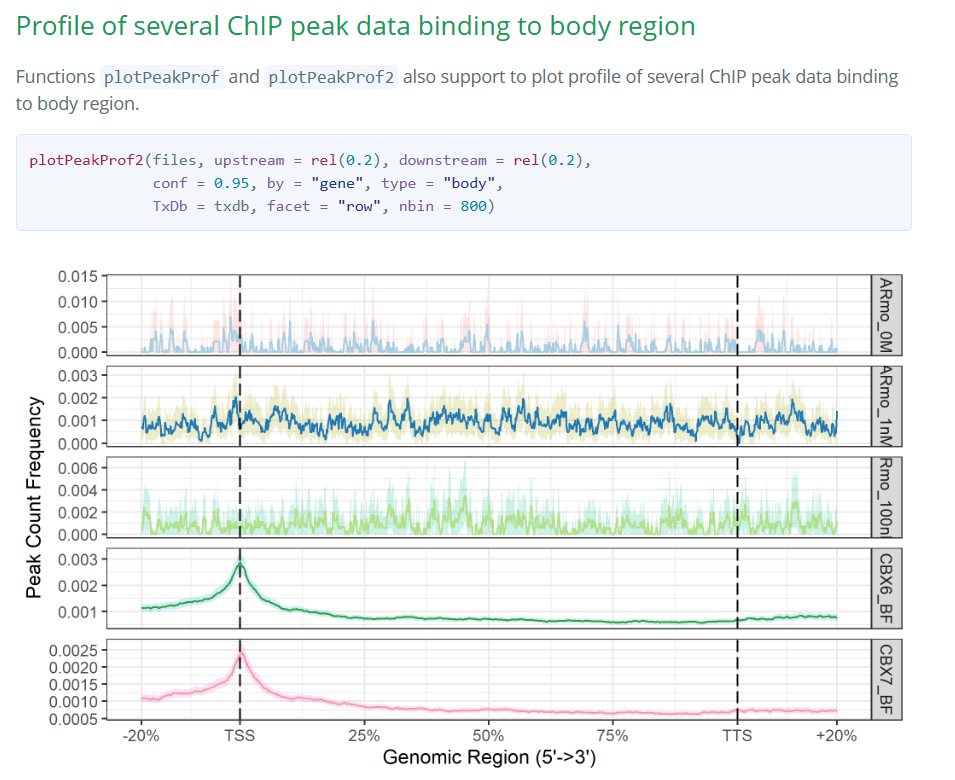
plotPeakProf2示例
plotPeakProf2,可以直接输入*summits.bed文件名(路径),通过指定 type = "body"将txdb文件中每一条序列的整个特定BioRegion作为一个窗口。
实践流程 & 代码
导入相关的包
1
2
3
4
5
|
library(ChIPseeker)
#install.packages("RMariaDB")
library(RMariaDB)
library(GenomicFeatures)
library(ggimage)
|
获取背景注释信息
GenomicFeatures包中有函数用来制作TxDb对象:
- makeTxDbFromUCSC: 通过UCSC在线制作TxDb
- makeTxDbFromBiomart: 通过ensembl在线制作TxDb
- makeTxDbFromGRanges:通过GRanges对象制作TxDb
- makeTxDbFromGFF:通过解析GFF文件制作TxDb
通过ucsc或者ensembl数据库ftp协议下载gff文件,如 ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/gff3/schiz
1
|
maize_TxDb <- makeTxDbFromGFF("E:/seq_project/work_data/Zma_B73_V4.gff3")
|
整段peak注释画图代码
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
|
bed_data_path <- "E:/seq_project/work_data/summits_bed"
orgs <- c("ear_1", "ear_2", "shoot_1", "shoot_2", "tassel")
plot_path <- "E:/seq_project/R包peak注释/plot"
for (org in orgs){
bed_files <- list.files(paste(bed_data_path, org, sep = "/"), pattern = "\\.bed$", full.names = TRUE)
bed_names <- gsub("_summits.*", "", basename(bed_files))
names(bed_files) <- bed_names
bed_files <- as.list(bed_files)
#依次读取bed文件
for (bed_file in bed_files){
peak <- readPeakFile(bed_file)
peakAnno <- annotatePeak(peak, tssRegion=c(-2000, 2000), TxDb=maize_TxDb)
pdf(file = file.path(plot_path,"pie", org, paste(basename(bed_file), "_pie", ".pdf", sep = "")))
plotAnnoPie(peakAnno)
dev.off()
pdf(file = file.path(plot_path,"pie", org, paste(basename(bed_file), "_upset", ".pdf", sep = "")))
p <- upsetplot(peakAnno, vennpie=TRUE)
print(p)
dev.off()
}
pdf(file = file.path(plot_path,"PeakProf", paste0(org, "genebody_PeakProf", ".pdf")))
p_1 <- plotPeakProf2(bed_files, upstream = 1000, downstream = 1000,
conf = 0.95, by = "gene", type = "body", nbin = 800,
TxDb = maize_TxDb, facet = "row")
print(p_1)
dev.off()
}
|
其他常规可视化
CS6: ChIP数据可视化 (guangchuangyu.github.io)
参考
CS6: ChIP数据可视化 (guangchuangyu.github.io)
CS4:关于ChIPseq注释的几个问题 (qq.com)
ChIPseeker: an R package for ChIP peak Annotation, Comparison and Visualization (bioconductor.org)