deeptools_computeMatrix使用介绍
computeMatrix & plot
文件格式
-
BED (Browser Extensible Data),一种灵活的储存数据的格式,主要用来 储存基因组特征 (genomic features) 或注释信息(*.summits.bed) 。
BED格式可用于UCSC的Genome Browser可视化工具中
-
挖掘生物数据信息时,我们会将进行序列比对(未知的序列与已知的reference对比,从而找到未知序列中隐藏的信息),常见的序列比对的文件输出格式为sam和bam
Sequence Alignment Mapping (SAM) 格式包括两部分:1. 注释信息(header section)2. 比对结果(alignment section)
Binary Alignment/Map (BAM)是SAM格式的二进制压缩格式,这两种格式是序列比对时软件常用的数据格式

- sam/bam格式文件,就是把测序reads比对到参考基因组后的文件
- bam或者bed格式的文件主要是为了追踪我们的reads到底比对到了参加基因组的什么区域
deeptools功能
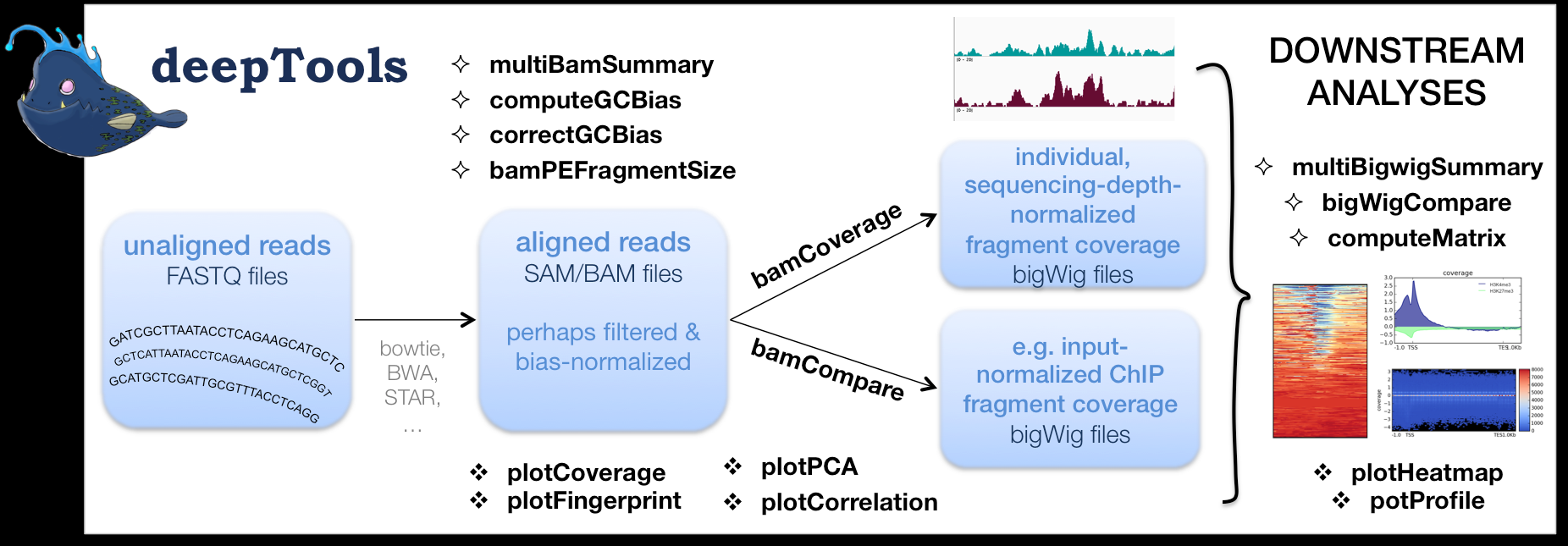
Deeptools 对 ChIP-seq 数据进行图形呈现
bam文件转换成bw文件格式
将bam文件转换为bigwig文件,这是一种压缩的二进制格式,可以快速加载和显示
|
|
得到的bw文件就可以送去IGV/Jbro wse进行可视化。 这里的参数仅使用了 -e/--extendReads和 -bs/--binSize即拓展了原来的read长度,且设置分箱的大小。其他参数还有
--filterRNAstrand {forward, reverse}: 仅统计指定正链或负链--region/-r CHR:START:END: 选取某个区域统计--smoothLength: 通过使用分箱附近的read对分箱进行平滑化
如果为了其他结果进行比较,还需要进行标准化,deeptools提供了如下参数:
--scaleFactor: 缩放系数--normalizeUsingRPKMReads: Per Kilobase per Million mapped reads (RPKM)标准化--normalizeTo1x: 按照1x测序深度(reads per genome coverage, RPGC)进行标准化--ignoreForNormalization: 指定那些染色体不需要经过标准化
computeMatrix
计算每个基因组区域的得分,并生成一个可与 plotHeatmap 和 plotProfiles 一起使用的中间文件。
|
|
computeMatrix 有两种主要的使用模式/参数

computeMatrix reference-point
reference-point 计算的基因组区域以某一个位置点作为相对参考点
--referencePoint {TSS,TES,center}
- region start (TSS)
- the region end(TES)
- the center of the region (默认为TSS)
computeMatrix scale-regions
scale-regions 计算的基因组区域为一段设定的区域长度(-b -a -m三个参数设置控制)
|
|
|
|
参考
computeMatrix — deepTools 3.2.1 documentation (test-argparse-readoc.readthedocs.io)
[生信资料 3] 生物信息学常见数据格式,汇总! - 知乎 (zhihu.com)