--shift:移动读取的中心位置。对于 ATAC-seq 数据,通常会设置为 -100,表示向 5’ 方向移动 100 bp
--extsize:设置读取的延伸大小。对于 ATAC-seq 数据,通常会设置为 200,表示每个读取都会向 3’ 方向延伸 200 bp
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
INFO @ Thu, 28 Mar 2024 15:32:03: #1 tag size is determined as 51 bps
INFO @ Thu, 28 Mar 2024 15:32:03: #1 tag size = 51.0
INFO @ Thu, 28 Mar 2024 15:32:03: #1 total tags in treatment: 27376162
INFO @ Thu, 28 Mar 2024 15:32:03: #1 user defined the maximum tags...
INFO @ Thu, 28 Mar 2024 15:32:03: #1 filter out redundant tags at the same location and the same strand by allowing at most 1 tag(s)
INFO @ Thu, 28 Mar 2024 15:32:03: #1 tags after filtering in treatment: 15521232
INFO @ Thu, 28 Mar 2024 15:32:03: #1 Redundant rate of treatment: 0.43
INFO @ Thu, 28 Mar 2024 15:32:03: #1 finished!
INFO @ Thu, 28 Mar 2024 15:32:03: #2 Build Peak Model...
INFO @ Thu, 28 Mar 2024 15:32:03: #2 Skipped...
INFO @ Thu, 28 Mar 2024 15:32:03: #2 Use 200 as fragment length
INFO @ Thu, 28 Mar 2024 15:32:03: #2 Sequencing ends will be shifted towards 5' by 100 bp(s)
INFO @ Thu, 28 Mar 2024 15:32:03: #3 Call peaks...
INFO @ Thu, 28 Mar 2024 15:32:03: #3 Pre-compute pvalue-qvalue table...
INFO @ Thu, 28 Mar 2024 15:33:16: #3 Call peaks for each chromosome...
INFO @ Thu, 28 Mar 2024 15:33:56: #4 Write output xls file... SRR5748809_peaks.xls
INFO @ Thu, 28 Mar 2024 15:33:56: #4 Write peak in narrowPeak format file... SRR5748809_peaks.narrowPeak
INFO @ Thu, 28 Mar 2024 15:33:57: #4 Write summits bed file... SRR5748809_summits.bed
INFO @ Thu, 28 Mar 2024 15:33:57: Done!
peakAnno<-annotatePeak(peak,tssRegion=c(-1000,1000),TxDb=maize_TxDb)#annotated peaks to gene and clusterProfilerlibrary(clusterProfiler)library(AnnotationHub)hub<-AnnotationHub()maize<-hub[["AH114308"]]entrez_gene_id<-read.csv("D:/motif_project/16.tss_maxmotif_GO/data/entrez_Zm_gene_v4.csv",header=T)gene_ids<-unique(as.data.frame(peakAnno)$geneId)%>%as.data.frame()colnames(gene_ids)<-"geneId"genes<-merge(gene_ids,entrez_gene_id,by.x="geneId",by.y="gene_name",all.x=T)%>%na.omit()ego<-enrichGO(gene=genes$ENTREZID,OrgDb=maize,keyType="ENTREZID",pAdjustMethod="BH",pvalueCutoff=0.8,qvalueCutoff=1,readable=TRUE)head(ego)write.table(ego,file=file.path(wd,paste0(basename(bed_file),"_GO.tsv")),sep="\t",quote=FALSE,row.names=FALSE)pdf(file=file.path(wd,paste0(basename(bed_file),"_GO_barplot.pdf")))dotp<-ggplot(ego,aes(GeneRatio,fct_reorder(Description,GeneRatio)))+geom_segment(aes(xend=0,yend=Description))+geom_point(aes(color=p.adjust,size=Count))+scale_color_viridis_c(guide=guide_colorbar(reverse=TRUE))+scale_size_continuous(range=c(2,10))+theme_minimal()+ylab(NULL)print(dotp)dev.off()
实验结果
数据预处理
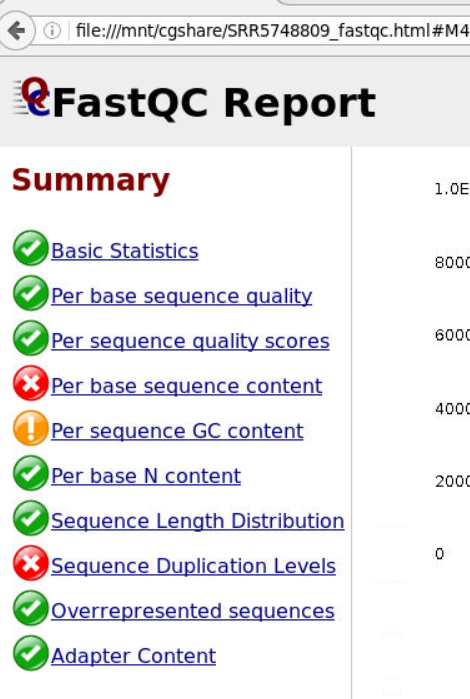
原始SRR5748809 QC
原始数据FastQC summary
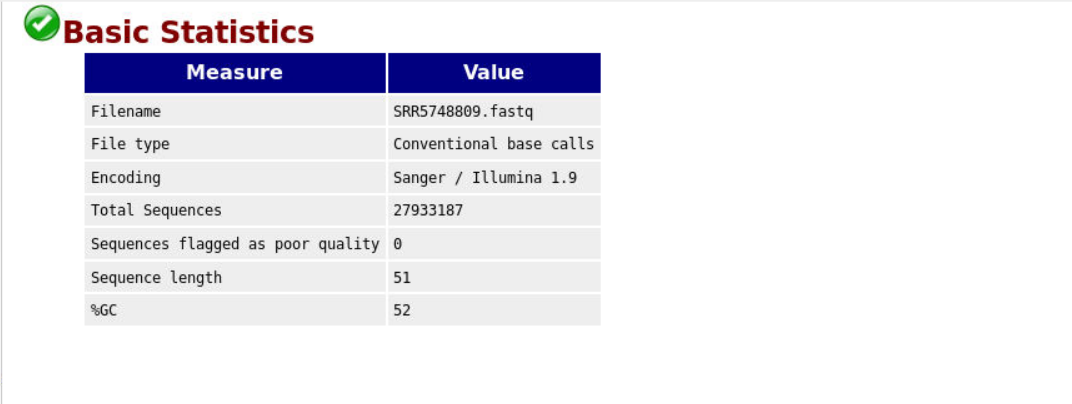
原始数据 Bsic Statistics
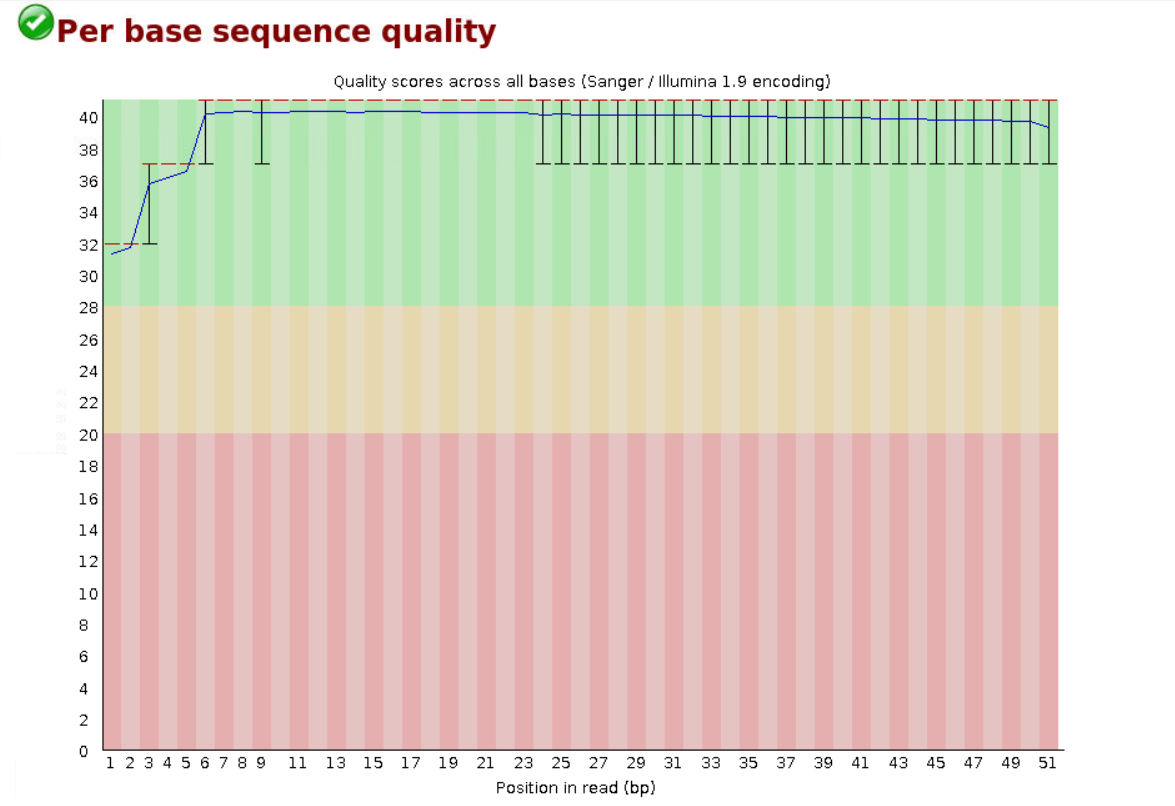
原始数据质量箱线图分布
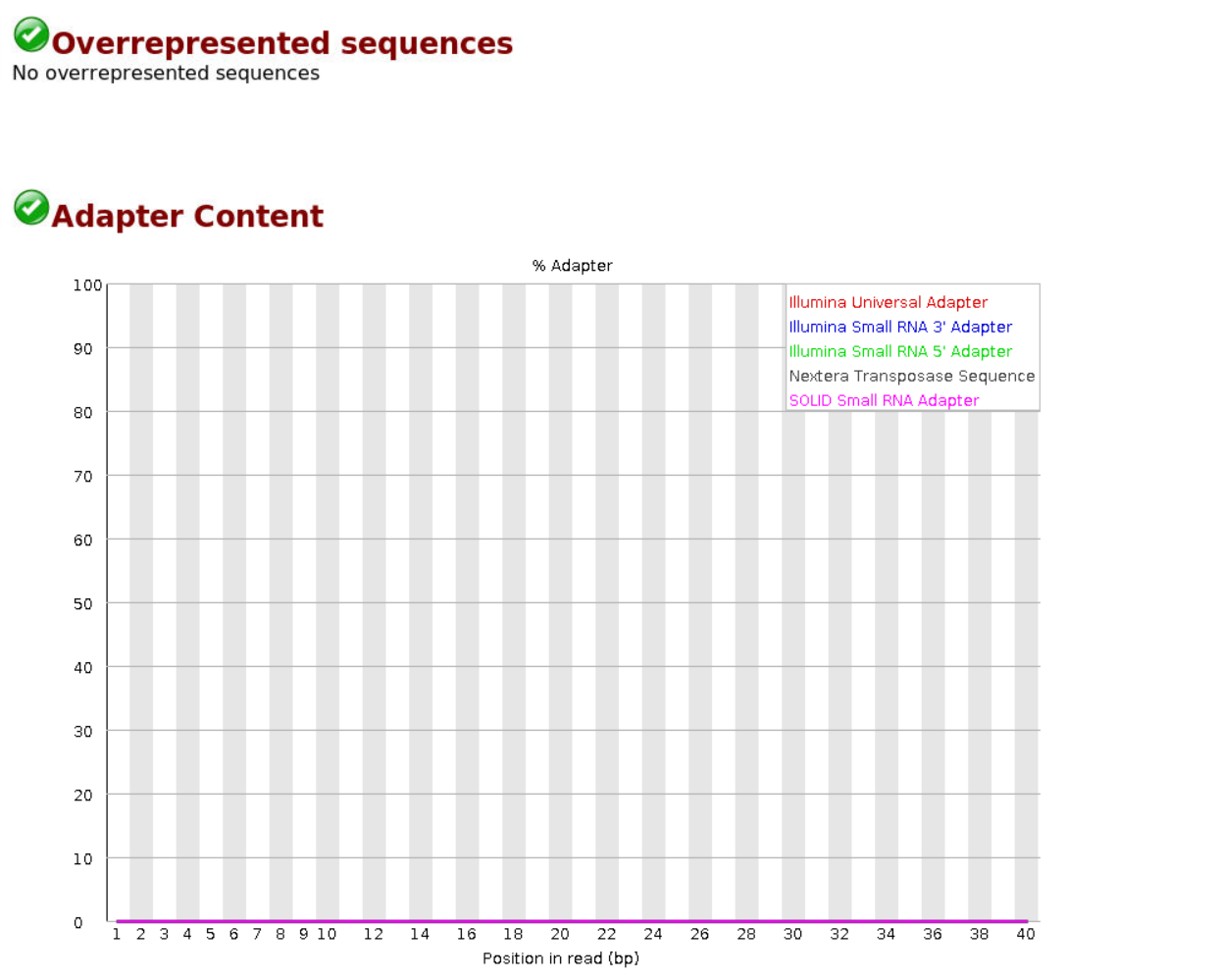
原始数据无过表达序列和接头
使用上述Trimmomatic命令剪切后,质量没有之前好
剪切数据质量summary
其中per base sequence content有些许改变,似乎可以通过 HEADCROP:14参数删除前14个碱基,但序列本身不是很长,便没有尝试
[bio_zxdai@bioinfo-01 align]$ cat flagstat.txt
27933221 + 0 in total (QC-passed reads + QC-failed reads)0 + 0 secondary
34 + 0 supplementary
0 + 0 duplicates
27376162 + 0 mapped (98.01% : N/A)0 + 0 paired in sequencing
0 + 0 read1
0 + 0 read2
0 + 0 properly paired (N/A : N/A)0 + 0 with itself and mate mapped
0 + 0 singletons (N/A : N/A)0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
peak calling
peak calling结果分析
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
INFO @ Thu, 28 Mar 2024 15:32:03: #1 tag size is determined as 51 bps INFO @ Thu, 28 Mar 2024 15:32:03: #1 tag size = 51.0 INFO @ Thu, 28 Mar 2024 15:32:03: #1 total tags in treatment: 27376162 INFO @ Thu, 28 Mar 2024 15:32:03: #1 user defined the maximum tags... INFO @ Thu, 28 Mar 2024 15:32:03: #1 filter out redundant tags at the same location and the same strand by allowing at most 1 tag(s) INFO @ Thu, 28 Mar 2024 15:32:03: #1 tags after filtering in treatment: 15521232 INFO @ Thu, 28 Mar 2024 15:32:03: #1 Redundant rate of treatment: 0.43 INFO @ Thu, 28 Mar 2024 15:32:03: #1 finished! INFO @ Thu, 28 Mar 2024 15:32:03: #2 Build Peak Model... INFO @ Thu, 28 Mar 2024 15:32:03: #2 Skipped... INFO @ Thu, 28 Mar 2024 15:32:03: #2 Use 200 as fragment length INFO @ Thu, 28 Mar 2024 15:32:03: #2 Sequencing ends will be shifted towards 5' by 100 bp(s) INFO @ Thu, 28 Mar 2024 15:32:03: #3 Call peaks... INFO @ Thu, 28 Mar 2024 15:32:03: #3 Pre-compute pvalue-qvalue table... INFO @ Thu, 28 Mar 2024 15:33:16: #3 Call peaks for each chromosome... INFO @ Thu, 28 Mar 2024 15:33:56: #4 Write output xls file... SRR5748809_peaks.xls INFO @ Thu, 28 Mar 2024 15:33:56: #4 Write peak in narrowPeak format file... SRR5748809_peaks.narrowPeak INFO @ Thu, 28 Mar 2024 15:33:57: #4 Write summits bed file... SRR5748809_summits.bed INFO @ Thu, 28 Mar 2024 15:33:57: Done!
tag size is determined as 51 bps:reads的大小为51个碱基对
filter out redundant tags at the same location and the same strand by allowing at most 1 tag(s):同一位置和同一链上的冗余标签被过滤掉
tags after filtering in treatment: 15521232:过滤后,处理的样本中有15521232个tags
0 + 0 paired in sequencing、0 + 0 read1、0 + 0 read2、0 + 0 properly paired (N/A : N/A)、0 + 0 with itself and mate mapped、0 + 0 singletons (N/A : N/A)、0 + 0 with mate mapped to a different chr、0 + 0 with mate mapped to a different chr (mapQ>=5): 这些统计信息都是关于配对的reads的,但在这个数据集中,所有的reads都是单端的,所以这些统计信息都是0
基因富集分析结论
GO富集分析结果显示了"protein-containing complex binding"、“cytoskeletal motor activity"和"microtubule motor activity"这三个GO项。这可能意味着在叶肉细胞中,有一些基因的调控区域是开放的,这些基因可能参与到蛋白质复合物的结合和细胞骨架的运动中。这些过程可能与叶肉细胞的一些重要功能有关,例如光合作用、物质运输等